Work
You can view some of my software projects at my GitHub page. Below is a small curated collection of the projects I am (and was) involved in.
Research Projects
• Jasper & Microbiome Maps
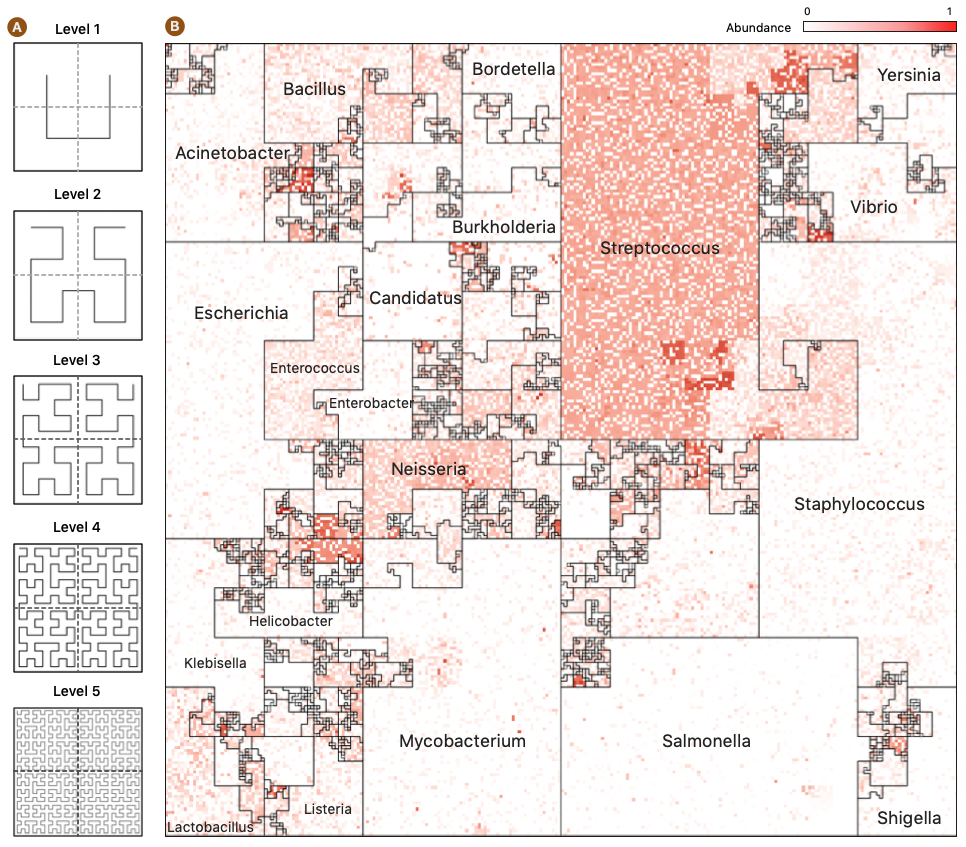

Microbiome Maps are visualizations of microbial community profiles, and they can be created with the Jasper software. Jasper is a tool for creating rich, interactive microbiome maps that lets you explore your metagenomic samples like never before. Jasper uses a Hilbert Curve to place genomes on an interactive canvas that can display thousands of genomes at once.

Flint is a metagenomics profiling pipeline that runs on Amazon Web Services (AWS), is built on top of the Apache Spark framework, and is designed for fast profiling of metagenomic samples against a large collection of reference genomes. Flint takes advantage of the Spark's built-in parallelism and streaming engine architecture to quickly map reads against a large reference collection of bacterial genomes. Our implementation relies distributing the alignment of millions of sequencing reads against a large collection of bacterial genomes. The genome collection is partitioned in order to distribute it across worker machines, and this allows the use of large collections of reference genomes.

Gene Expression Analysis in Xenograft samples by RNA-Seq and DNA Microarrays
Gene expression studies analyze how genes are "turned On" or "turned Off" in a given sample or condition, and there are many tools for doing this.
High-throughput mRNA sequencing, or RNA-Seq, is a great tool for studying gene expression in diseases such as cancer because it provides a more precise measurement than previous methods. In this research paper we studied how RNA-Seq compared with another tool, DNA Microarrays, for detecting gene expression changes in tumor and stromal cancer samples. We focused on gene models from the Consensus Coding Sequences (CCDS), and noted how both technologies behaved in our samples.

Detecting Bacterial Genomes in Metagenomic Samples
The study of how microbes affect human disease is an area I am very much interested in. In this research paper we developed a method that uses frequentist probabilities to detect bacterial strains in metagenomic samples — samples that are collected from anywhere: rivers, soil, humans, etc — using DNA sequencing data.
Our approach introduces artificial point mutations at the nucleotide level in each DNA sequencing read to define a test statistic for each genome in our reference database of bacterial genomes. Most of the methods out there have a detection resolution that goes as far as the Species-level, but our method achieved a Strain-level resolution.